|
|
|
|||||
|
||||||
MAH試點、進入ICH、關聯申報:倒逼原料藥企升級提速摘要:
醫藥網5月2日訊 在新政策環境及其產生的連鎖效應下,原料藥企的全面升級迫在眉睫。
2017年11月30日,原CFDA關于調整原料藥、藥用輔料和藥包材審評審批事項的公告(2017年第146號),要求有關企業或者單位可通過登記平臺按公告要求提交原料藥、藥用輔料和藥包材登記資料,獲得原料藥、藥用輔料和藥包材登記號,待關聯藥品制劑提出注冊申請后一并審評。公告發布前已獲得批準文號的原料藥、藥用輔料和藥包材相關登記要求,將在登記平臺建立后另行通知。
在登記平臺建立的過渡期,藥審中心在門戶網站以表格方式對社會公示“原料藥登記數據”“藥用輔料登記數據”“藥包材登記數據”。以原料藥為例,原料藥登記資料主要內容包括基本信息、生產信息、特性鑒定、原料藥的質量控制、對照品、藥包材、穩定性等。
同年12月4日,原CFDA辦公廳公開征求《原料藥、藥用輔料及藥包材與藥品制劑共同審評審批管理辦法(征求意見稿)》意見(以下簡稱“征求意見稿”)。這意味著原料藥、藥用輔料及藥包材(以下簡稱“原輔包”)關聯審評審批制度的序幕即將拉開。
上市許可持有人責任大
[趨勢] 制劑質控從嚴,對原輔包企提出新要求
根據“征求意見稿”,藥品上市許可持有人承擔制劑質量的主體責任,建立以制劑為核心、以原輔包為基礎的質量管理體系。這就要求藥品制劑上市許可持有人建立的質量管理體系能涵蓋制劑全生命周期的質量管理,對制劑所用的原輔包質量能夠有效追溯,文件管理中必須要明晰原輔包來源、批次、生產、質控和變更情況。
藥品制劑上市許可持有人需要和原輔包企業一同簽署“原輔包企業保證持續穩定地供應符合制劑質量的原輔包產品的長期供貨/質量保證協議”,原輔包企業提交必要信息以便評估和控制由原輔包引入制劑的質量風險。藥品制劑與原輔包不是同一申請人的,藥品制劑申請人的申報資料還要提供原輔包上市許可持有人或者企業的授權使用書,這要求藥品制劑上市許可持有人在原輔包的關聯合同中要有相關訴求。
藥品上市許可持有人還要對原輔包開展供應商審計,從制劑全生命周期的質量管理新理念出發,審計將要在研發階段前啟動。審計的內容一般包括現場GMP考核、工廠廠商資質審核和質量體系文件審核、產品注冊文件和年度報告。值得關注的是,研發階段一些輔料所需的供應量比較低,輔料供應商未必愿意接受審計;此外,如果原輔包企業覺得藥品上市許可持有人有盜取資料的風險,也有可能會拒絕合作。
隨著一致性評價法規對制劑的體外溶出速率、生物利用度和穩定性要求不斷提高,粒徑大小若是影響溶出度、溶解度、生物利用度,制劑生產、穩定性、含量均勻度,以及產品外觀的關鍵因素,通常都要制定粒徑限度要求。在這種情況下,國內的制劑企業通常選擇將壓力往上游傳遞,要求原料藥生產企業加強粒徑控制,特別是直接混合原輔料壓片工藝制劑對應的原料藥,以往的過篩控制已不能滿足制劑生產企業的需求。
此外,根據“征求意見稿”,同一原料藥生產企業供不同給藥途徑制劑使用且質量存在差別的同一原料藥,應當按不同登記號登記;給藥途徑相同、生產工藝相近,僅晶型、粒徑等質控要求不同的原料藥,應當在同一登記號下對不同工藝、晶型、粒徑進行分類并編號。這將有利于原料藥生產企業供貨不同粒徑控制需求的制劑企業。
ICH法規受注重
[趨勢] 倒逼合作原料藥廠質量升級
中國進入ICH后,原料藥也要參考ICH原料藥開發和制造管理原則。目前CDE的法規與規章欄目“ICH指導原則”的質量指導原則已發布了不少原料藥相關的中文版,如《Q1A(R2):新型原料藥和藥品的穩定性測試》《Q1B: 穩定性測試: 新型原料藥和藥品的光穩定性測試》《Q1D:新型原料藥和藥品穩定性測試的交叉和矩陣設計》《Q3A(R2): 新型原料藥中的雜質問題》《Q6A: 質量規格:新原料藥和藥品的檢驗程序和可接收標準:化學物質》和《Q7: 原料藥GMP指南》等與原料藥相關的指導原則。
ICH制度影響最大的是國內已有批文的原料藥廠家,下游制劑生產企業一致性評價質量提升壓力,將倒逼已合作的原料藥廠家的質量體系進行升級。例如雜質譜的全面研究,包括降解雜質、遺傳毒性雜質和元素雜質的研究;殘留溶劑中有無應避免的溶劑,應限制的溶劑又無超過規定 PDE(the Permitted Daily Exposure,允許日接觸量);起始物料的審計與管理制度等。
關聯申報“試用期”
[趨勢] 歐美原料供貨商將回流
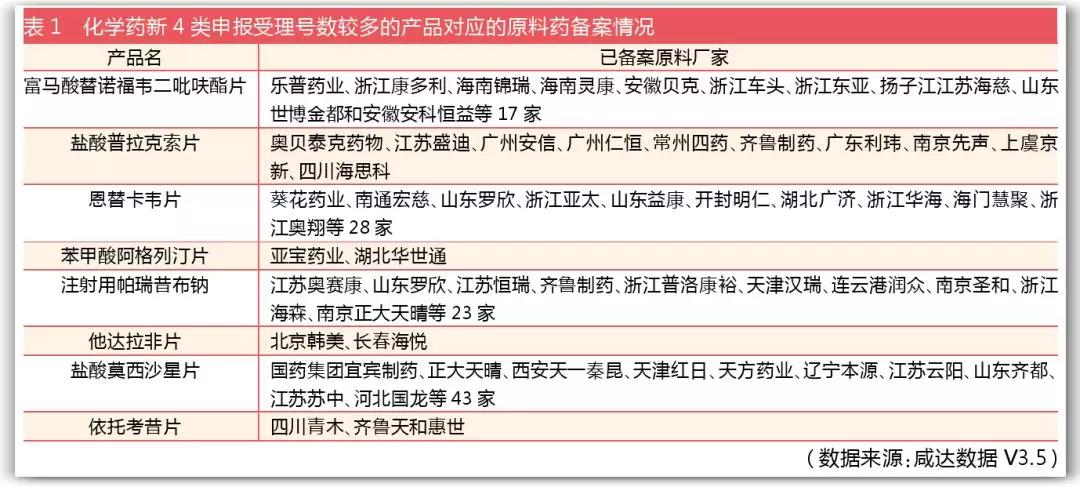
截至2018年4月23日,共有2037條原料藥信息備案。咸達數據V3.5整理了化學藥新注冊分類辦法4類的申報受理號數較多的產品對應的原料藥廠家備案情況后發現,除了富馬酸替諾福韋二吡呋酯片原研廠家香港吉立亞科學有限公司備案的原料藥狀態為“已有上市制劑使用該原料”,新4類的其他制劑對應的原料藥暫無“單獨申報且已通過技術審評的原料,尚未與制劑進行共同審評”或“已登記但尚未通過技術審評的原料,正在與關聯制劑共同審評”狀態,個別產品為“已登記但尚未通過技術審評的原料,尚無關聯制劑申報”狀態。由此可見,原料藥和制劑的關聯情況暫不能從數據庫中獲悉。
一直致力于歐美市場的中國原料藥生產廠家,以往由于價格和注冊法規的原因,絕少供貨國內生產廠家。關聯審評政策出臺后,由于一致性評價的剛性需求,這些企業有可能回流國內參與原料藥市場的競爭。制劑企業在選擇此類企業的時候,需要了解過往現場核查中主要發現了哪些缺陷,還要了解國內外注冊法規要求的不同點,例如國內原料藥CTD文件要求原料藥也要做包材相容性的研究。
為了保證一致性評價的注冊進度,制劑企業選擇原料藥廠家時,會選擇在美國有DMF、歐盟CEP申請,并且通過歐美GMP認證和現場核查的生產廠家。目前供貨的主要有歐洲、印度和中國的生產企業。
過了一致性評價后,制劑上市許可持有人更換原料藥生產企業是否還需要重新做生物等效性試驗法規暫未明確。根據歐美的法規,制劑上市許可持有人根據其對原料藥的標準重新選擇經濟性更好的原料藥企業只需要做三批驗證即可。關聯申報制度下,原料藥備案平臺中“已有上市制劑使用該原料”對應的非原研廠家的原料藥生產商就可以參與原料藥供應競爭,以往國內由于生產批件所造成的原料藥壟斷情形會減少,相應地,技術壁壘和專利所導致的市場供應不足將會越來越多出現。
 |
|
Copyright ?2015 廣東穗康醫藥有限公司
粵ICP備15022662號(粵)
-技術支持:信息管理部|聯系我們
|